Cientistas desenvolvem ferramenta de software inteligente para avaliação de risco químico
Nos últimos anos, os modelos de aprendizado de máquina se tornaram cada vez mais populares para avaliação de risco de compostos químicos. No entanto, eles são frequentemente considerados “caixas pretas” devido à sua falta de transparência, levando ao ceticismo entre toxicologistas e autoridades regulatórias. Para aumentar a confiança nesses modelos, pesquisadores da Universidade de Viena propuseram identificar cuidadosamente as áreas do espaço químico onde esses modelos são fracos. Eles desenvolveram uma ferramenta de software inovadora ('MolCompass') para esse propósito e os resultados dessa abordagem de pesquisa acabaram de ser publicados no prestigiado Journal of Cheminformatics.
Ao longo dos anos, novos produtos farmacêuticos e cosméticos foram testados em animais. Esses testes são caros, levantam preocupações éticas e muitas vezes não conseguem prever com precisão as reações humanas. Recentemente, a União Europeia apoiou o projeto RISK-HUNT3R para desenvolver a próxima geração de métodos de avaliação de risco não animais. A Universidade de Viena é membro do consórcio do projeto. Os métodos computacionais agora permitem que os riscos toxicológicos e ambientais de novos produtos químicos sejam avaliados inteiramente por computador, sem a necessidade de sintetizar os compostos químicos. Mas uma questão permanece: quão confiáveis são esses modelos de computador?
É tudo uma questão de previsão confiável
Para abordar essa questão, Sergey Sosnin, um cientista sênior do Pharmacoinformatics Research Group da Universidade de Viena, focou na classificação binária. Nesse contexto, um modelo de aprendizado de máquina fornece uma pontuação de probabilidade de 0% a 100%, indicando se um composto químico é ativo ou não (por exemplo, tóxico ou não tóxico, bioacumulativo ou não bioacumulativo, um ligante ou não ligante para uma proteína humana específica). Essa probabilidade reflete a confiança do modelo em sua previsão. Idealmente, o modelo deve estar confiante apenas em suas previsões corretas. Se o modelo for incerto, dando uma pontuação de confiança em torno de 51%, essas previsões podem ser desconsideradas em favor de métodos alternativos. Um desafio surge, no entanto, quando o modelo está totalmente confiante em previsões incorretas.
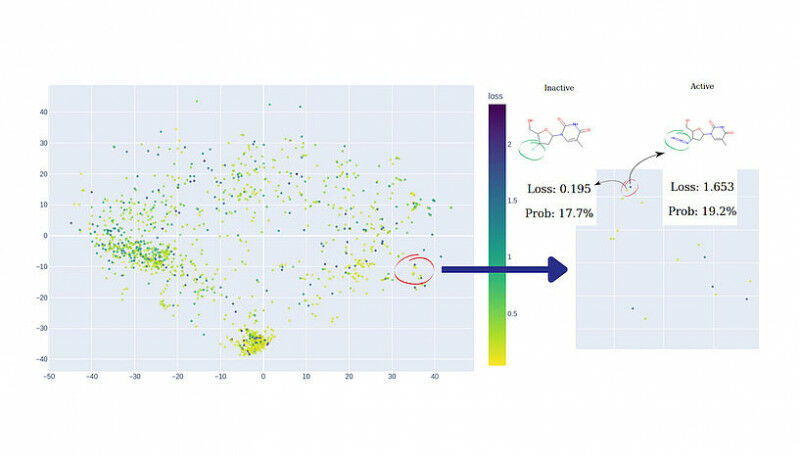
“Este é o verdadeiro cenário de pesadelo para um toxicologista computacional”, diz Sergey Sosnin. “Se um modelo prevê que um composto não é tóxico com 99% de confiança, mas o composto é realmente tóxico, não há como saber que algo estava errado.” A única solução é identificar áreas de “espaço químico” – abrangendo possíveis classes de compostos orgânicos – onde o modelo tem “pontos cegos” com antecedência e evitá-los. Para fazer isso, um pesquisador que avalia o modelo deve verificar os resultados previstos para milhares de compostos químicos, um por um – uma tarefa tediosa e propensa a erros.
Superando esse obstáculo significativo
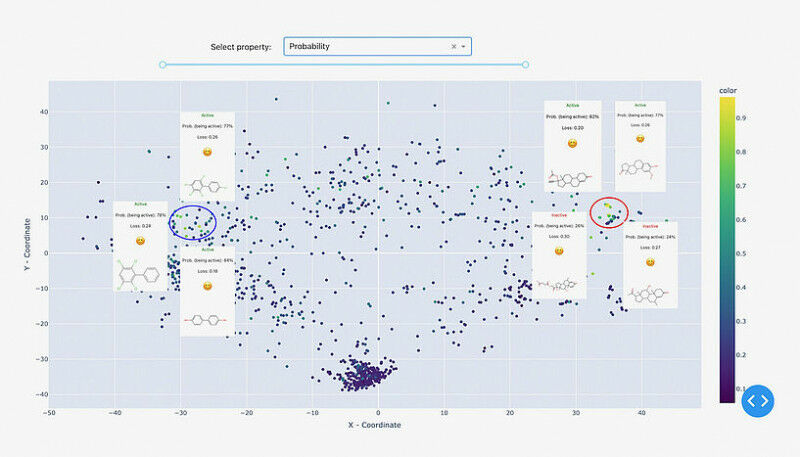
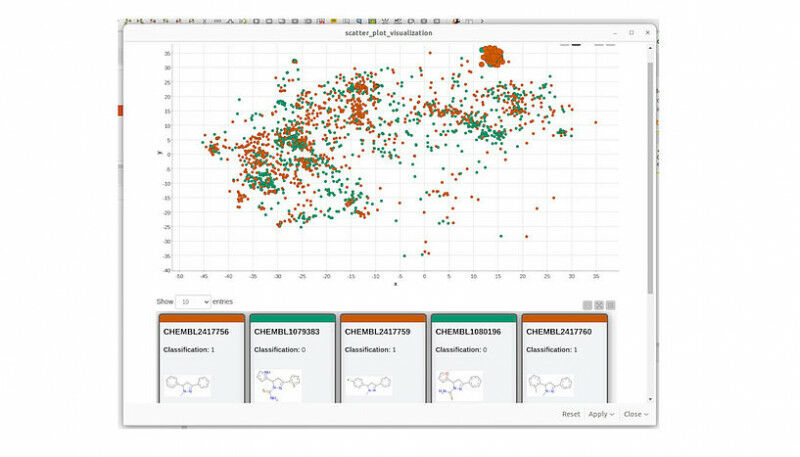
“Para auxiliar esses pesquisadores”, continua Sosnin, “nós desenvolvemos ferramentas gráficas interativas que exibem compostos químicos em um plano 2D, como mapas geográficos. Usando cores, destacamos os compostos que foram previstos incorretamente com alta confiança, permitindo que os usuários os identifiquem como aglomerados de pontos vermelhos. O mapa é interativo, permitindo que os usuários investiguem o espaço químico e explorem regiões de preocupação.”
A metodologia foi comprovada usando um modelo de ligação do receptor de estrogênio. Após análise visual do espaço químico, ficou claro que o modelo funciona bem para, por exemplo, esteroides e bifenilas policloradas, mas falha completamente para pequenos compostos não cíclicos e não deve ser usado para eles.
O software desenvolvido neste projeto está disponível gratuitamente para a comunidade no GitHub. Sergey Sosnin espera que o MolCompass leve químicos e toxicologistas a uma melhor compreensão das limitações dos modelos computacionais. Este estudo é um passo em direção a um futuro onde os testes em animais não são mais necessários e o único local de trabalho para um toxicologista é uma mesa de computador.
Publicação original:
S. Sosnin: MolCompass: multiferramenta para navegação no espaço químico e validação visual de modelos QSAR/QSPR. Journal of Cheminformatics.
DOI: 10.1186/s13321'024 -00888-z
Fig. 1: A demonstração do MolCompass ilustra como um toxicologista computacional pode identificar áreas preocupantes do espaço químico. Usando nosso software, o toxicologista pode identificar regiões onde o modelo sob investigação prevê incorretamente a atividade com alta confiança. C: Sergey Sosnin Fig. 2: Uma ilustração da busca por penhascos de modelo. No lado esquerdo do mapa químico, dois pontos estão próximos, mas exibem cores contrastantes. Uma investigação mais aprofundada dessa observação peculiar revela que, embora esses dois compostos compartilhem um alto grau de similaridade estrutural, eles exibem atividades opostas, representando um desafio que o modelo não consegue abordar efetivamente. Os dados visualizados são: Conjunto de dados de ligantes de estrogênio. C: Sergey Sosnin Fig. 3: Dois clusters foram atribuídos com alta confiança pelo modelo de referência. O cluster mais denso à esquerda representa derivados de esteroides, enquanto o cluster direito, menos definido, inclui bifenilos policlorados e polifenóis. Os dados visualizados são: Conjunto de dados de ligantes de estrogênio. C: Sergey Sosnin Fig. 4: Uma captura de tela da visualização KNIME do espaço químico usando o nó MolCompass KNIME. C: Sergey Sosnin